La Ocronosis alkaptonurica es una rara enfermedad autosómica recesiva del metabolismo de la tirosina y fenilalanina, caracterizada por una triada de orinas oscuras (Alkaptonuria), coloración negruzca de tejidos conectivos (ocronosis) y degeneración articular en adultos jóvenes (artritis ocronotica)1, por deficiencia del la 1,2 dioxigenasa homogentísica, produciendo la acumulación del Acido Homogentísico (HGA). De tratamiento poco efectivo tanto con Vitamina C como con Nitisinone, aunque este último inhibe síntesis del HGA. Se hace el reporte de una paciente femenina de 53 años, con gonartrosis bilateral severa a predominio izquierdo, sin antecedentes patológicos conocidos, que durante la artroplastia se evidenciaron lesiones negruzcas en todo el cartílago articular, por lo que se tomaron muestras para anatomía patológica que reportaron lesiones sugestivas de artritis ocronótica. Posteriormente se evidencia orinas oscuras a la exposición al aire libre, artropatía degenerativa no limitante en esqueleto axil y parentesco de consanguinidad tanto de sus abuelos como de su padres. Se hace este reporte por ser el primer caso registrado en la literatura nacional, además de su excepcional frecuencia.
Palabras clave: Alkaptonuria, ocronosis, artritis ocronótica, ácido homogentísico.
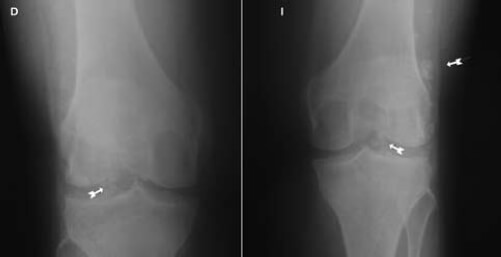
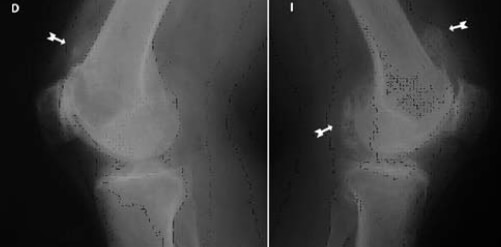
Paciente femenina de 53 años de edad, mestiza, evaluada en la consulta de Traumatología de Hospital Dr. Luis Salazar Domínguez, por cuadro de gonalgia progresiva y bilateral de 2 años de evolución a predominio izquierdo, que se exacerba a la marcha, limitando la misma, con sensación de bloqueo de ambas rodillas y que en los últimos meses no responde al tratamiento convencional con AINES, y sin antecedentes de importancia. Al examen físico articular de ambas rodillas se evidencia en un ligero aumento de volumen periarticular a predominio izquierdo; en la rodilla izquierda con flexión limitada a 130°, extensión limitada a 160°, y la rodilla derecha con flexión limitada a 100° y extensión máxima de 180°; resto del examen físico general sin evidencia de alteraciones. En las proyecciones radiográficas de ambas rodillas en apoyo anteroposteriores y laterales se aprecia una artrosis grado IV (figura nº 1) femororrotuliana y femorotibial, con abundantes calcificaciones ovaladas dispersas sugestivas de osteocondromatósis primaria, con predominio de las lesiones en la rodilla izquierda. Perfil reumatológico negativo.
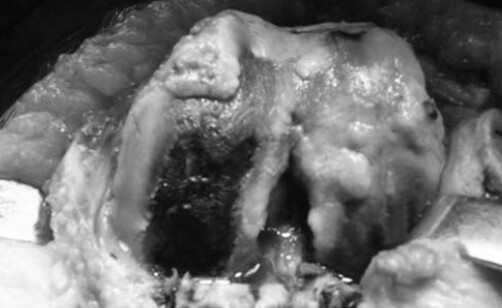
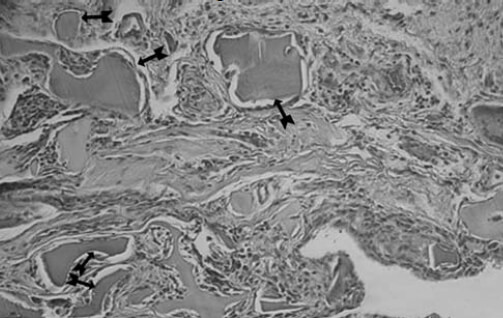
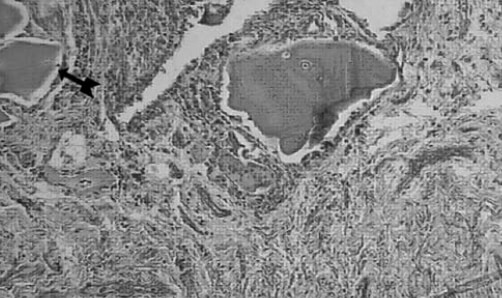
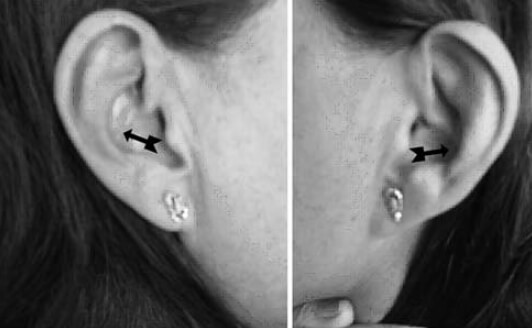
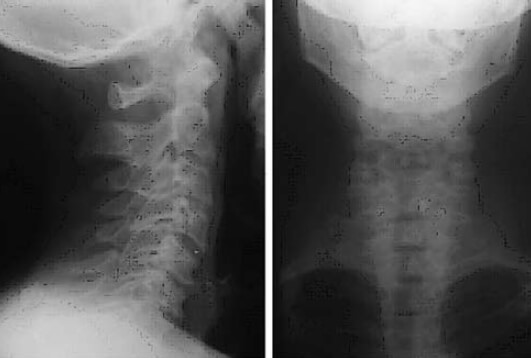
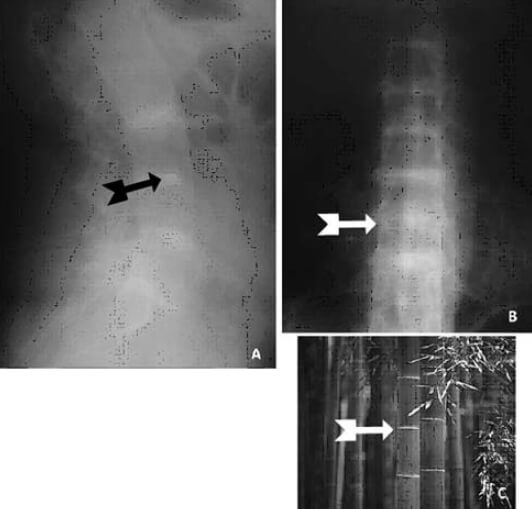
Se realiza cirugía de reemplazo articular, inicialmente de la rodilla izquierda con el diagnóstico de Osteoartritis Degenerativa Primaria; durante el acto quirúrgico se evidencia superficie articular irregular, friable, fragmentado, con áreas de coloración negruzca y bordes externos grisáceos, (figura nº 2), los meniscos y ligamentos cruzados también se aprecian teñidos de negro, intraarticularmente se extraen abundantes cuerpos libres ovalados compatibles con osteocondromatosis primaria. Los cortes realizados evidencian que la coloración negruzca abarca todo el espesor cartilaginoso (figura nº 3). Se toman muestras de los cortes y cuerpos libres y fijan en formol al 10%, enviándose al Departamento de Patología Ósea del Instituto de Anatomía Patológica Dr. José Antonio O’Daly, (muestra B-000676-08) que reporta osteoartritis crónica con fragmentación del cartílago articular y pigmentación de la matriz cartilaginosa (cartílago negro) compatible con ocronosis y sinovitis crónica con reacción giganto-celular tipo cuerpo extraño (figuras nº 4 y 5). En vista del reporte anatomopatológico se reevalúa a la paciente en busca de estigmas. Se evidencia nexos de consanguinidad directos (primos hermanos) tanto de sus abuelas y padres, uroanálisis donde después de 1 hora se evidencia oscurecimiento de la muestra en contacto con el aire, con un viraje total a negro en aproximadamente 6 horas (figura nº 6), ambos pabellones auriculares con zona de color pardo oscuro (figura nº 7), estudios radiológicos de la columna vertebral con artrosis severa en columna cervical (figura nº 8), en columna dorso lumbar se aprecia artrosis severa con osteopenia acentuada de los cuerpos vertebrales y calcificación de los discos intervertebrales dando una apariencia de “palo de bambú” (signo de palo de bambú) (figura nº 9), en ambas articulaciones coxofemorales se aprecia artrosis grado II, el resto de la evaluación no evidencia alteraciones.


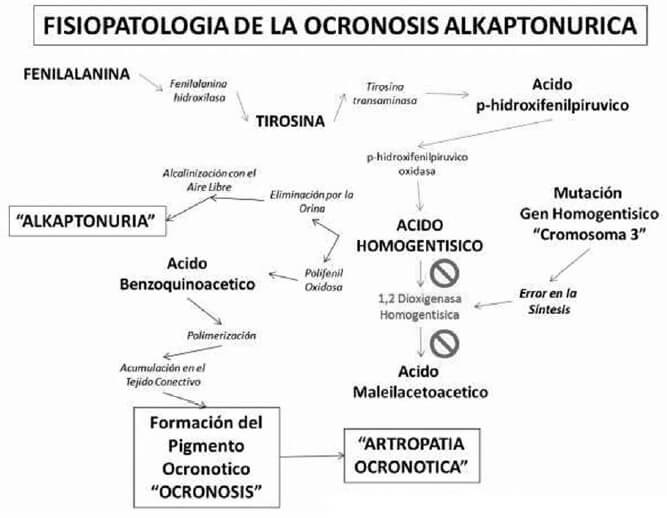
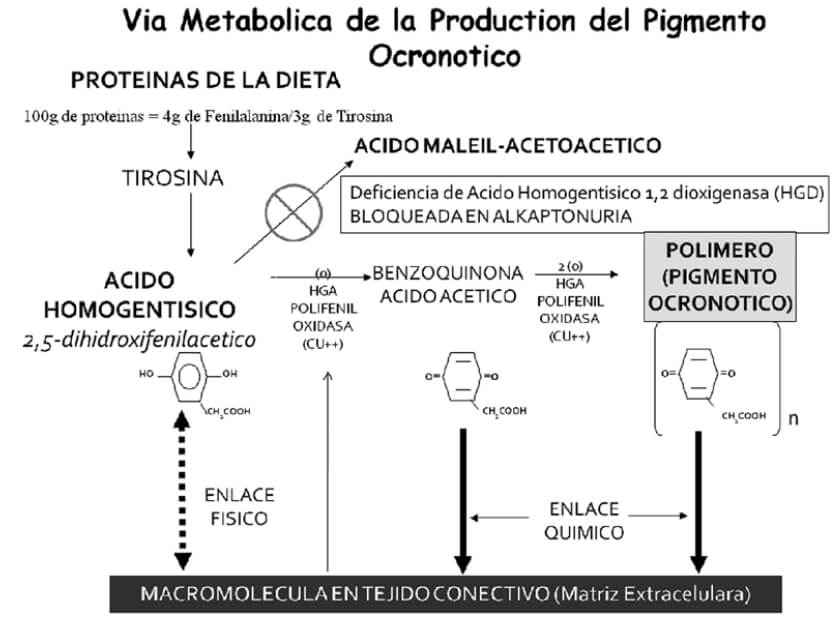
La Ocronosis alkaptonurica es una rara enfermedad hereditaria autosómica recesiva, que origina una alteración en la vía metabólica de los aminoácidos fenilalanina y tirosina (figura nº 10). Tal afectación se debe a mutaciones en el gen HGO (homogentísico), ubicado en el brazo largo del cromosoma 32, en las regiones 3q21-q23, lo cual produce errores en la síntesis parcial o completa de la oxidasa del ácido homogentísico (1,2 dioxigenasa homogentisico), enzima solitaria presente en el hígado y en el riñón, responsable del catabolismo del Acido Homogentísico (HGA) a Acido Maleilacetoacético; siendo así la primera enfermedad en donde se demuestra su transmisión de padres a hijos siguiendo las leyes Mendelianas # 2035001,3, descrito por Alchibald Garrod hace 100 años, el cual introduce por primera vez el termino de “inborn error of metabolism” y propone a la alkaptonuria como parte de estas alteraciones3,4,5,6. Su incidencia a nivel mundial va desde 1/250.000, 1/500.000 hasta 1/1.000.0001,2,3,4, con países como República Dominicana y Slovakia en donde la incidencia es menor 1/20.0001,3,4. Esta entidad se encuentra clasificada dentro de las enfermedades huérfanas “orphan disease”4.
Las primeras descripciones de esta enfermedad datan de 1500 A.C. observadas en momias egipcias1,3, Escribonius 1584 y Schenck en 1609 hacen descripciones de los primeros casos3,6, para 1859 se introduce por primera vez el termino “alkapton” para referirse a las orinas de los pacientes con tales características, pero no es hasta 1908 que Garrod lo describe como un error hereditario del metabolismo “Inborn Errors of Metabolism”; para 1959 Neubauer describe la vía metabólica de la tirosina y en el mismo año, Cervenansky y col. hacen una descripción detallada de la enfermedad8, hasta que en 1996 Fernandez-Canon y col. clonan el Gen HGO7.
La alkaptonuria es el resultado de la acumulación en la orina del HGA no catabolizado por la deficiencia de la 1,2 dioxidasa homogentísica, por lo que en contacto con el oxígeno atmosférico se alcaliniza, desencadenando una reacción de oxidación y polimerización del HGA, que como consecuencia hace que la orina progresivamente vaya oscureciendo su color original de amarillo a pardo oscuro y negro en el transcurso de pocas horas, siendo este proceso aun mas rápido cuando el pH de la orina es alcalino8. Por esto el fenómeno recibió el nombre de Alkapton (termino híbrido entre la palabra árabe que designa álcali y la griega que indica absorber), que posteriormente recibe el nombre de alkaptonuria. Este fenómeno se demuestra rápidamente en el laboratorio, cuando hace reaccionar la orina que contiene grandes cantidades de HGA con soluciones Fehling ó Benedict, reacción demostrada en 18591,8. También se ha notado que los restos de orina impregnados en la ropa interior blanca al momento de ser lavada con jabones con pH alcalino, tiñe de gris la tela; este fenómeno preocupa a las madres de los niños con alkaptonuria. A pesar de que el oscurecimiento de la orina es relativamente rápido, no es raro que este problema pase inadvertido tanto para las madres como para los pacientes adultos, por lo precoz del descarte de la muestra.
La confirmación diagnostica de la presencia de grandes o pequeñas cantidades del HGA en orina es mediante la demostración del ácido por espectrografía enzimática1,3,4,8. El diagnóstico también puede ser confirmado utilizando cromatografía líquida de alta presión, método que cuantifica la cantidad de HGA y sus derivados1, por lo que este método es utilizado para la monitorización al momento de administrar tratamiento. La excreción del HGA en orina es usualmente alta, con valores de 0,4 a 12 g. de este compuesto excretado diariamente1,3, y siendo muy pocas las cantidades encontradas en el plasma con un rango de 3 a 20 ug. por ml. día, valores indetectables en pacientes que no padecen de la enfermedad3. La ocronosis es el resultado de la acumulación del HGA en el tejido conectivo en forma de pigmento ocronótico, dando una coloración negruzca y/o pardo oscura a tales tejidos, de esta particularidad nace el termino de ocronosis, descrito inicialmente por Virchow8. Este pigmento se origina de la oxidación y polimerización del HGA en forma de benzoquinona en la matriz extracelular de los tejidos conectivos (figura nº 8). En el cartílago de los mamíferos, la polifenil oxidasa, cataliza la oxidación del HGA en pigmento ocronótico depositándose en gránulos. Sin embargo, gránulos intracelulares se encuentra presentes en los condrocitos; no se conoce si el pigmento depositado y su unión a los componentes de la matriz extracelular ocurren primariamente a nivel intracelular o extracelular4.
Muchos son los aparatos y sistemas afectados, principalmente el sistema músculo esquelético. Las rodillas, hombros, caderas y columna vertebral son las partes más afectadas. Los depósitos ocronóticos ocasionan la fragilidad y fragmentación del cartílago. A nivel ocular se produce una pigmentación parda escleral y epiescleral por delante de la inserción de los músculos rectos, sobre todo lateral y medio. En el limbo corneal puede verse una pigmentación subepitelial de aspecto de “gota de aceite”, este pigmento puede también depositarse sobre cicatrices oculares antiguas9. En el sistema cardiovascular la incidencia de la enfermedad es alta12; las válvulas aórtica y mitral son las más afectadas debido a los cambios degenerativos, calcificación y valvulitas, que en los casos severos ameritan reemplazo valvular de urgencia1. Los polvos ocronóticos pueden causar la calcificación o endurecimiento de las válvulas cardíacas. También, los depósitos de los pigmentos pueden llevar a la formación de placas ateroescleróticas (manchas firmes en las arterias) que contienen colesterol y grasa. El síndrome coronario agudo con infarto del miocardio es la causa común de muerte1. En el sistema genitourinario, en los varones, la próstata es el órgano más afectado. Los depósitos de los pigmentos conducen a la formación de cálculos en la próstata. En el sistema respiratorio, son comunes fuertes depósitos de la pigmentación en el cartílago de la laringe, la tráquea, y en los bronquios. En la piel, los efectos son más notorios en las zonas expuestas al sol y donde se sitúan las glándulas sudoríparas. La piel adopta una coloración de manchas negruzcas. El sudor produce manchas de color marrón en la ropa. También se evidencian lesiones en la dentadura, en el sistema nervioso central (cerebro y médula espinal), y en los órganos endocrinos10.
La artropatía ocronótica es la principal manifestación clínica de esta enfermedad, comportándose como una osteoartritis degenerativa secundaria a la afectación del cartílago, que aparece generalmente en individuos jóvenes con una media de inicio en la tercera década, afectando sobre todo a las articulaciones vertebrales y de carga. Esto se caracteriza generalmente como una artropatía de comienzo insidioso con sintomatología no marcada. Los primeros síntomas articulares aparecen en la columna vertebral, manifestándose como una sensación de rigidez a nivel lumbosacro, pero que generalmente no va acompañada de dolor como se aprecia en los procesos inflamatorios, a pesar de lo avanzado de los cambios patológicos observados en las proyecciones radiológicas. En casos tempranos se evidencia solo aplanamiento de las curvaturas fisiológicas, evolucionando a cifosis a partir del sexto u octavo año después de la aparición de los primeros síntomas8, los cambios son mas acentuados en la sexta, séptima y octava vertebra torácica; otras de las alteraciones que se observan es el estrechamiento de los espacios intervertebrales, con posterior calcificación de los discos intervertebrales y una marcada osteopenia de los cuerpos vertebrales (signo del palo de bambú) (figura nº 9). Tales cambios osteoporóticos son el resultado de una hiperplasia reactiva secundaria, que deforma el hueso y causa su destrucción8. A partir de este punto se desencadena rigidez del la columna toracolumbar con la anquilosis, zonas de cifosis, lordosis y escoliosis, también se atrofia de la musculatura paravertebral. Las claves en el diagnóstico diferencial entre la artropatía ocronótica y la artritis anquilosante, son las evidencias clínicas y radiológicas en la columna cervical; en los pacientes con ocronosis las alteraciones radiológicas no se correlacionan con las manifestaciones clínicas, a diferencia de los procesos reumatológicos donde tanto las alteraciones radiológicas son directamente proporcionales a la sintomatología. Los pacientes con artropatía ocronótica presentan menos limitación funcional y dolor que aquellos que presentan osteoartritis de la columna cervical. Sin embargo, la artropatía ocronótica evoluciona rápida y extensivamente, causando rigidez toracolumbar temprana, comparado con los procesos de osteoartritis. La artropatía ocronótica a nivel de la articulación de la rodilla es la más frecuente1,4,8,10, (seguida de la articulación coxofemoral), con compromiso bilateral, de rápida evolución, en donde los cambios degenerativos observados en las proyecciones radiológicas convencionales son abundantes, tales como la perdida de la línea articular con esclerosis de las articulaciones femororrotuleana y femorotibial, presencia de abundantes osteofitos marginales, y calcificaciones ovaladas de bordes irregulares, tendencia a la deformidad en valgo; macroscópicamente una superficie irregular con zonas negruzcas, bordes grisáceos azulados, tendencia a la formación de cuerpos libres intraarticulares tipo osteocondromatosis primaria. La limitación funcional es progresiva a pesar de los cambios radiológicos, con tendencia a limitación a la flexión, que en casos severos ameritan reemplazo articular total11. Aunque la incidencia es mayor en hombres, la incidencia de la enfermedad es igual para ambos sexos12. A nivel del resto de las articulaciones de carga se evidencian cambios similares a los de la rodilla, a diferencia de la mayor tendencia a la formación de pseudoquistes y calcificaciones de las inserciones tendinosas a predominio de la articulación gleno humeral8.
Anatomopatológicamente, los hallazgos al microscopio incluyen cambios degenerativos del cartílago, pigmentación difusa de la matriz cartilaginosa y pigmentación de la membrana sinovial. Los cambios óseos son menos severos, remodelación ósea y mineralización.
Las opciones terapéuticas para esta afectación se basan principalmente en medidas paliativas en pro de disminuir la acumulación del HGA en los tejidos conectivos y así enlentecer el proceso degenerativo a nivel articular y el resto de las alteraciones en los tejidos diana, aunque poco efectivos, uno de los esquemas iniciales es a nivel dietético, con la disminución en la ingesta de proteínas, ya que se estima que por cada 100 g. de proteínas de la dieta, 4 g. son de Fenilalanina y 3 g. de Tirosina, esto reduce de alguna manera el sustrato para la síntesis de HGA. La administración de vitamina C vía oral, con esquemas de 0,5 g. VO BID hasta de 1 g. VO BID, solo ha demostrado la disminución de la excreción urinaria de las benzoquinonas del acido acético, pero no reduce la excreción de HGA en la orina3. Entre otros de los esquemas de tratamiento aun en fase experimental, es el tratamiento farmacológico con Nitisinone, un compuesto que busca la reducción en la formación de HGA, debido a que este fármaco inhibe aunque de forma reversible la 4-hidroxifenilpiruvato dioxigenasa, por lo que no se formar el HGA a causa de la inhibición en el catabolismo de su sustrato predecesor. El nitisinone ha sido aprobado por la FDA para el tratamiento de la tirosinemia tipo 1, una enfermedad hepática hereditaria, fatal. El enfoque terapéutico en los casos avanzados, se basa en diagnosticar a tiempo la diversidad de alteraciones en los tejidos diana, en la búsqueda del diagnóstico de las probables complicaciones que se puedan generar. Cuando la artropatía en las articulaciones de carga es limitante y severa, el remplazo articular es la opción ideal, lo cual ayuda al paciente a mejorar la calidad y estilo de vida.
Consideramos importante la publicación de este caso, debido a lo raro de la enfermedad, lo extraordinario de su aparición. Después de una revisión bibliográfica de la literatura nacional, no encontramos otro caso publicado y reportado sobre esta patología, que a pesar su rareza, es importante que la comunidad médica venezolana tenga el conocimiento de su existencia y saber cuáles son sus complicaciones y las medidas terapéuticas que se tomaran para el tratamiento y seguimiento de los pacientes afectados.