

 ,
Manuel Brito2 ,
Isver Bracho3 ,
Firás Souki Chmeit4
,
Manuel Brito2 ,
Isver Bracho3 ,
Firás Souki Chmeit4
Fecha de recepción: 21 de marzo de 2014. Fecha de aceptación: 09 de septiembre de 2016.
El “síndrome de Karsch-Neugebauer”, conocido comúnmente como ectrodactilia, es una malformación congénita de las extremidades, caracterizada por la presencia de hendiduras en pies y manos que se produce por agenesia e hipoplasia de los huesos metacarpianos, metatarsianos y falanges. Es un desorden genético raro, con una incidencia de 1:90.000 – 100.000 nacimientos vivos, sin relación con el sexo y con prevalencia mundial desconocida. El diagnóstico es clínico, y se realiza al momento del nacimiento, aunque se puede realizar el diagnóstico prenatal mediante ultrasonografía. El principal objetivo del tratamiento es conseguir una mano que sea funcional y permita el agarre de objetos y la realización de actividades básicas, y conseguir un pié plantígrado que permita el apoyo, que sea funcional para la marcha y que permita el uso de calzado. Presentamos el caso un paciente femenino de 9 años con síndrome de Karsch-Neugebauer, que fue manejada quirúrgicamente por nuestro servicio con buenos resultados clínicos y funcionales. Presentamos su evolución a corto, mediano y largo plazo. Rev Venez Cir Ortop Traumatol, 2017, Vol 49 (2): 75-80.
Palabras clave: Deformidades Congénitas del Pie, Deformidades Congénitas de la Mano, Pie, Huesos del Pie, Genética.
Nivel de Evidencia: 4
The “Karsch-Neugebauer syndrome”, commonly known as ectrodactyly, is a congenital malformation of the extremities, characterized by the presence of cracks in feet and hands that results from agenesis and hypoplasia of the metacarpals, metatarsals and phalanges. It is a rare genetic disorder with an incidence of 1:90.000 - 100,000 live births, without regard to sex and prevalence worldwide unknown. The diagnosis is clinical, and is performed at birth, but can perform prenatal diagnosis by ultrasonography. The main goal of treatment is to get a hand that is functional and allows the gripping of objects and the basic activities, and achieve a plantigrade foot that allows support to be functional for walking and allow the use of footwear. We report the case of a female patient 9 years with Karsch-Neugebauer syndrome, which was managed surgically by our service with good clinical and functional results. We present its short and long term evolution. Rev Venez Cir Ortop Traumatol, 2017, Vol 49 (2): 75-80.
Key words: Congenital Foot Deformities, Congenital Hand Deformities, Foot, Foot Bones, Genetics.
Level of evidence: 4
Autor de correspondencia: Manuel Brito, email: [email protected]
Conflictos de interés: Los autores declaran que no existen conflictos de interés. Este trabajo fue realizado con recursos propios sin subvenciones.
El “síndrome de Karsch-Neugebauer” (SKN), conocido como ectrodactilia, es una malformación congénita de las extremidades, caracterizada por la presencia de hendiduras en pies y manos que se produce por agenesia e hipoplasia de 4los huesos metacarpianos, metatarsianos y falanges. La palabra ectrodactilia proviene del griego “Ektrona” (aborto) y “Dactylos” (Dedo), literalmente (aborto de los dedos), y fue utilizado por primera vez por Cruveilhier en 1842. Otros nombres utilizados son “Split hand and foot” o “lobster claw hand and foot” (inglés) o “manos y pies hendidos” (español) (1,2,11,12).
Se considera un desorden genético raro, con incidencia de 1:90.000 – 100.000 nacimientos vivos, y no está relacionada al sexo, con prevalencia mundial desconocida (1,2,4,11).
La etiología principal de esta anomalía es genética, resaltando las bases hereditarias de trasmisión; los casos esporádicos, que son muy poco comunes; y los agentes teratogénicos, como los derivados del acido retinoico, el cadmio, etanol, cafeína, cocaína, ácido valproico, entre otros (7-9).
Genéticamente, es una enfermedad autosómica dominante con penetrancia genética variable, que en la mayoría de los casos es incompleta (2). Y se han descrito algunos casos del tipo autosómico recesivo (5,11).
Se genera principalmente por mutaciones cromosómicas del tipo estructural, asociándose a mutaciones en cinco diferentes locus (SHFM1 en 7q21.3, SHFM2 en Xq26, SHFM3 en 10q24, SHFM4 en 3q27 y SHFM5 en 2q31) (2,5,8).
Estas alteraciones afectan el proceso de inducción de la cresta ectodérmica apical sobre el mesodermo en la quinta semana de la fase embrionaria del desarrollo, previo al momento de la formación de los rayos digitales, lo cual modifica el proceso de diferenciación de los dedos posteriormente (2, 4, 9, 11).
El diagnostico es clínico, y se realiza al momento del nacimiento, aunque se puede realizar el diagnóstico prenatal mediante ultrasonografía desde el primer trimestre de la gestación (2,3,5,6,16).
Se han descrito dos formas clínicas, una forma “aislada” y la “ectrodactilia, displasia ectodérmica y hendidura de labio y paladar” mejor conocida como síndrome EEC (en inglés) que también se asocia a otros defectos faciales, genitourinarios y oculares (1).
Según Abraham y cols (14), se clasifican en 3 tipos, de acuerdo a su presentación clínica y sobre la cual implementan las recomendaciones terapéuticas. El tipo 1, la variedad más frecuente, se observa una hendidura o deficiencia del radio central sin separación de los radios medial o lateral; El tipo 2, con hendidura central profunda que alcanza los huesos del tarso; y El tipo 3, con ausencia completa del segundo al cuarto rayo (1,4,9,12,14).
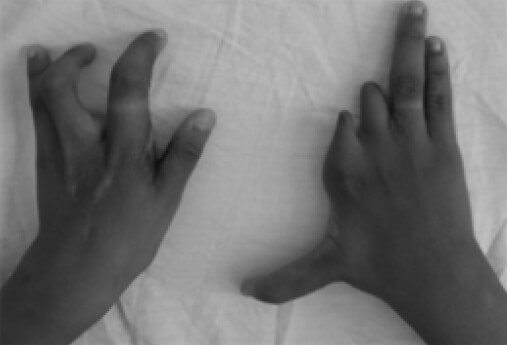
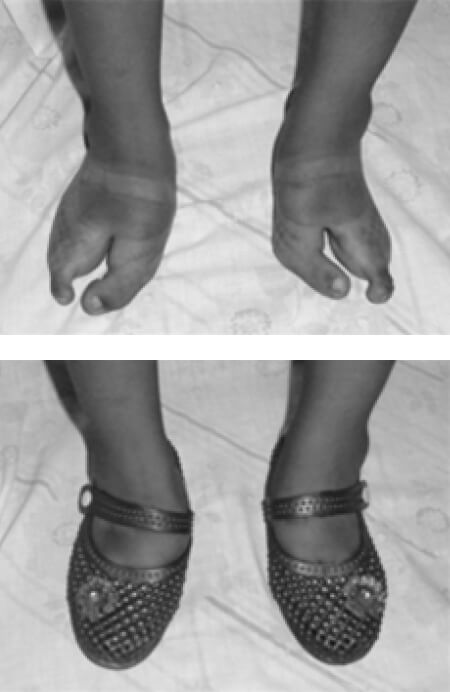
Paciente femenino de 8 años, natural y procedente de La Fría, Estado Táchira, quien presenta deformidad congénita de manos y pies. En la mano izquierda se evidencia sindactilia del dedo anular y medio, y en derecho se aprecia agenesia de los dedos medio e índice. En los pies se evidencia hendidura central con sindactilia bilateral del segundo, tercero, cuarto y quinto dedo, y hallux valgus bilateral (Figura 1 y 2).
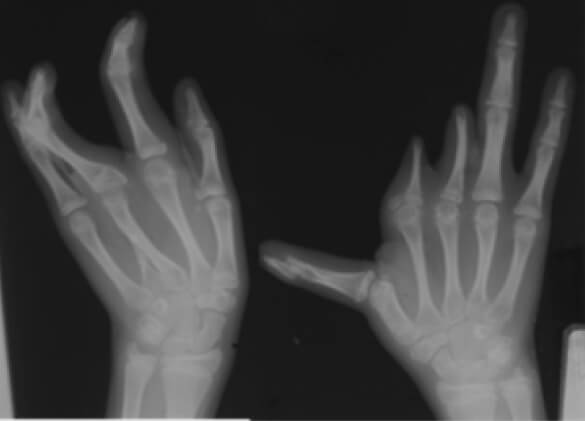
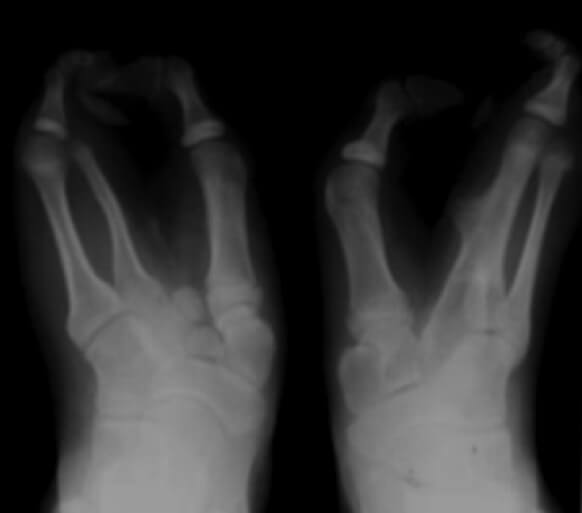
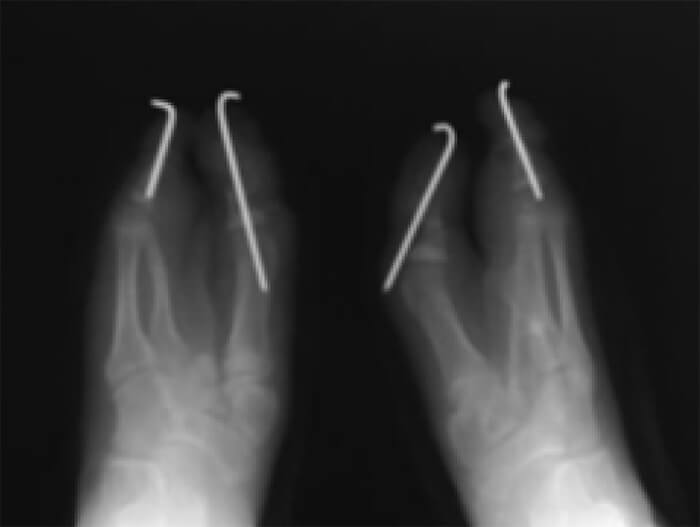
En la radiografía simple se observa, en la mano izquierda, fusión del segundo y tercer metacarpiano, con ausencia de las falanges del dedo medio. En la derecha, agenesia del primer metacarpiano, con segundo a quinto sin alteraciones, agenesia de la falange proximal del índice y medio, y ausencia de sus respectivas falanges media y distal. Mientras que en ambos pies se evidencia la ausencia de múltiples metatarsianos y falanges (Figura 3 y 4).
No se encontraron otras alteraciones dismórficas y la valoración antropométrica se encontraba dentro de límites normales. No se encontraron otras alteraciones físicas o sistémicas al momento del examen. La niña era estudiante de primaria, con buen desempeño escolar y desenvolvimiento para su edad. Segunda hija de un matrimonio no consanguíneo, producto de embarazo simple a término no complicado, obtenida mediante parto eutócico sin complicaciones o eventualidades perinatales. No hay historia de casos clínicos similares en ninguno de los lados materno o paterno. Sólo refiere un caso de agenesia total de todas las falanges medias y distales en la abuela materna.
Se procedió al tratamiento no quirúrgico con cinchas metatarsales y separadores de dedos como medidas preoperatorias para ambos pies.
Se procedió a la resolución quirúrgica mediante la supresión de la hendidura central en pies y manos con sindactilias de tipo lineal, primero se operaron las manos y dos años después los pies. Adicionalmente en el pié, se realizó la realineación de las falanges mediante osteotomía y osteodesis con alambres de Kirschner para corrección del hallux valgus (Figura 5).
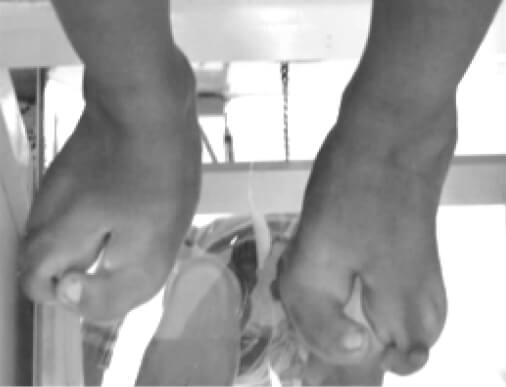
En el post operatorio, los pies se manejaron con cinchas metatarsales durante 1 mes. La evolución post operatoria fue favorable. Se mantuvo un seguimiento de 2 años donde hemos visto que sus pies y manos se comportan adecuadamente, desde el punto de vista mecánico, sus pies se comportan satisfactoriamente, permitiendo el uso de calzado. La escala visual análoga del dolor reportó un puntaje de 10/10 puntos a los 2 años (Figura 6 y 7).

Las expectativas de los pacientes con SKN con respecto a su tratamiento son altas, y el mismo debe estar dirigido a mejorar la calidad de vida, buscando el mejoramiento morfo funcional de pies y manos. El principal objetivo del tratamiento es conseguir una mano que sea funcional y permita el agarre de objetos y la realización de actividades básicas, y conseguir un pié plantígrado que permita el apoyo, que sea funcional para la marcha y que permita el uso de calzado.
Sin embargo, el impacto psicológico que tiene esta malformación en los pacientes, desde el punto de vista estético y funcional, convierten a su tratamiento en un verdadero reto para el cirujano ortopedista. Dicho tratamiento debe ser estratégico, bien planificado e individualizado. La inspección clínica y la realización de los diferentes estudios imagenológicos, como radiografías, tomografía computarizada y resonancia magnética, revelan datos importantes, como huesos atróficos y fusionados, además de las agenesias e hipoplasias óseas (4,11).
El tratamiento de esta anomalía incluye tanto el manejo conservador no operatorio como el operatorio, pero este último va a depender de que tan acentuada sea la malformación (11). De acuerdo a la clasificación de Abraham y col (14), las tipo 1, pueden no requerir tratamiento o por el contrario una cirugía
sobre partes blandas logrando la sindactilización; las tipo 2, requieren cirugías reconstructivas en pacientes jóvenes; y las tipo 3, que se caracterizan por la ausencia completa del 1 y 4 radio, no requerirían cirugías del antepie (14,15). El tratamiento no operatorio, incluye la utilización de distintos tipos de ortesis, separadores de dedos y cinchas metatarsales, que permiten una mejor alineación del pie (11).El manejo operatorio, debe ser adaptado a cada paciente, y puede ir desde el cierre de partes blandas, amputación de dedos, hasta reconstrucciones anatómicas, funcionales y estéticas, planteándose también el uso de prótesis (1, 4).
Epeldegui (13) sostiene que, con el manejo operatorio, se puede conseguir un apoyo plantígrado del pie y a su vez, alcanzar notables mejoras estéticas. Cuando se trate de huesos intercalares, recomienda su resección para prevenir la progresión de la divergencia de los radios del pie, lo que dificulta el uso del calzado normal (4).
Javed y Sultán (12), sugieren que las deformidades en manos y pies pueden ser tratadas quirúrgicamente para proveerles función y apariencia.
Según Allie y cols (11) se deben aplicar medidas conservadoras que permiten una mejor alineación del pie como preparación para la cirugía, como el uso de cinchas y ortesis. Luego de este tratamiento pre operatorio, y de la cirugía, también se deben tomar medidas post operatorias para evitar la desviación natural hacia la hendidura original.
Cienfuegos y cols. (4) recomiendan que, en casos de deformidades desiguales, se opere primero el que produzca mayor limitación y molestias al paciente.
Nuestro caso presenta ciertos aspectos particulares, su forma clínica de presentación es la aislada, es decir, que no se encuentra asociada a otras alteraciones clínicas o fisiológicas, la cual es la forma menos común del SKN; Esta forma clínica, por lo general sigue el patrón autosómico dominante de herencia con una alta penetrancia, sin embargo, en nuestro caso el patrón parece ser autosómico recesivo como caso único en la familia.
Para el manejo del pié hendido, recomendamos el tratamiento no operatorio como preparación para el tratamiento quirúrgico y mantenerlos en el post operatorio.
El tratamiento quirúrgico realizado y el adecuado manejo post operatorio, permitieron mejorar la función y apariencia, alcanzando los objetivos primarios del manejo del SKG.